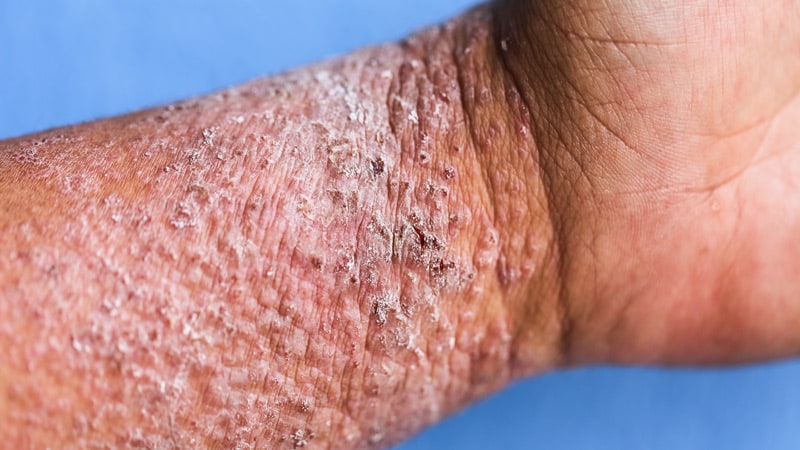
Cuma günü, Avrupa İlaç Ajansı'nın (EMA) İnsan Ürünleri Tıbbi Ürünleri Komitesi (CHMP), orta ila şiddetli atopik dermatitli yetişkinlerin tedavisi için IL-13 inhibitörü tralokinumab (Adtralza) için olumlu bir görüş yayınladı ( AD) sistemik tedaviye uygun olanlar.
Yeni görüş, Avrupa Komisyonu'nun tralokinumabın Avrupa Birliği genelinde kullanım için yetkilendirilip yetkilendirilmeyeceğine karar vermesinden önceki son aşamaları temsil etmektedir. Nihai karar önümüzdeki birkaç ay içinde verilmelidir.
Nihayetinde izin verilirse, tralokinumab, AD belirtilerini ve semptomlarını yönlendiren anahtar bir faktör olan IL-13 sitokini hedefleyen ilk onaylanmış tamamen insan monoklonal antikoru olacaktır. Tralokinumab daha önce IL-13'ü yüksek afinite ile hedeflediğini ve ardından enflamatuar cilt hastalığı ile ilişkili semptomları iyileştirdiğini göstermiştir.
EMA, geçen Haziran ayında tralokinumab için pazarlama başvurusunu kabul etti. Pazarlama uygulamasının yanında sunulan, ECZTRA 1, 2 ve 3 temel randomize, plasebo kontrollü çalışmalardan elde edilen verilerdir.
ECZTRA denemelerinde, tralokinumab ile tek başına veya topikal kortikosteroidlerle tedavi, açık veya hemen hemen berrak derinin Araştırmacı Küresel Değerlendirme skorunda önemli gelişmeler ve Egzama Alanı ve Şiddet İndeksi skorunda (EASI -75). Bu çalışmalarda tralokinumabın güvenliği, plasebo ile bildirilenle karşılaştırılabilir düzeydedir.
Açık etiketli uzatma çalışması ECZTEND'den elde edilen ara veriler de tralokinumab ile tedavinin orta ila şiddetli AD'li yetişkin hastalarda kalıcı etkinlik ile ilişkili olduğunu göstermiştir. Bu çalışmadaki hastalar daha önce ECZTRA 1 ve 2 ebeveyn denemelerine kaydolmuş ve 2 yıla kadar IL-13 inhibitörü almışlardır. Bu denemenin verileri kısa süre önce American Academy of Dermatology Virtual Meeting Experience (AAD VMX) 2021 sırasında sunuldu.
Avrupa Komisyonu'nun nihai kararına kadar, orta ila şiddetli AD'li yetişkinlerde tralokinumab kullanımına yönelik Pazarlama Yetkilendirme Başvurusu İzlanda, Norveç ve Lihtenştayn'a ek olarak tüm Avrupa Birliği üye devletlerinde geçerli olacaktır. İlaca ilişkin diğer yasal başvurular şu anda dünya çapında çeşitli ülkelerden sağlık yetkilileriyle devam etmektedir.
Temmuz ayında, ABD Gıda ve İlaç Dairesi, yetişkinlerde orta ila şiddetli AD'nin tedavisi için tralokinumab için bir Biyolojik Lisans Başvurusunu (BLA) kabul etti. Önemli ECZTRA 1, 2 ve ECZTRA 3 faz 3 çalışmalarından elde edilen veriler de BLA ile birlikte FDA'ya gönderildi.
Reçeteli İlaç Kullanıcı Ücreti Yasası uyarınca FDA, bu yılın ikinci çeyreğinde yetişkin AD endikasyonu için ABD'de tralokinumab yetkisi verip vermeme konusunda nihai bir karar vermeyi bekliyor.
Daha fazla haber için Medscape'i Facebook , Twitter , Instagram , YouTube ve LinkedIn'de takip edin
Recommended for you






You must be logged in to post a comment Login
Leave a Reply
Yorum yapabilmek için oturum açmalısınız.